
Qu’est ce que le rétinoblastome ? Quelles sont les causes de cette pathologie ? Quels sont les symptômes pour l’identifier ? Quels sont les traitements adéquats ? Découvrez ce que dit la science sur cette maladie. Attention cependant à ne pas confondre les informations fournies à des recommandations médicales.
Sommaire
Présentation du rétinoblastome
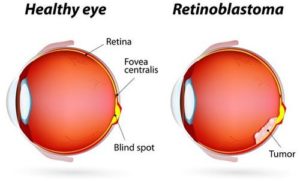
Le rétinoblastome est le cancer intraoculaire le plus couramment rencontré chez les jeunes enfants de moins de 5 ans. Il est le plus souvent unilatéral, c’est-à-dire n’affectant qu’un œil mais peut également être bilatéral dans environ 25% des cas. La tumeur se développe au niveau de la rétine qui n’est autre que l’organe photosensible des yeux. En l’absence d’une prise en charge précoce, elle peut rapidement conduire à diverses complications telles qu’un décollement rétinien, une hémorragie, la perte totale de la vue voire la mort suite à une dissémination du cancer et à la survenue de métastases.
Il s’agit d’une pathologie génétique qui touche environ une naissance sur 20.000 dans les pays européens. Plus de 50% des cas sont diagnostiqués avant l’âge de 2 ans mais d’autres cas semblent ne se manifester que beaucoup plus tard. Certains ne sont même découverts qu’après l’âge de 10 ans. Cette affection représente, en outre, près de 3% des cas de tumeurs infantiles qui peuvent être rencontrés.
Causes du rétinoblastome
Ce type de cancer intraoculaire affectant les jeunes enfants est une pathologie génétique qui peut provenir soit :
– De l’hérédité, par transmission autosomique dominante. Dans tel cas, il suffit qu’un seul des parents transmet le gène muté à l’enfant pour que celui-ci soit sujet à la pathologie. Cependant, la mutation acquise par voie héréditaire n’altère que l’un des deux allèles du gène responsable de la tumeur. La manifestation des symptômes nécessite pourtant un changement simultané des deux allèles. Une deuxième mutation est donc nécessaire pour qu’il y ait activation de la tumeur. Celle-ci ne survient qu’au niveau des cellules rétiniennes ;
– D’une mutation somatique du gène responsable de la maladie qui a lieu dans les cellules de la rétine. Les deux allèles sont dans tel cas simultanément altérés. La véritable cause de cette mutation n’est pas encore cependant clairement identifiée.
Les formes bilatérales de la tumeur sont toujours dues à une prédisposition génétique héritée d’un des parents et concernent près du quart des cas. Pour ce qui sont des formes unilatérales, elles surviennent dans 15% des cas pour la transmission héréditaire et le reste soit environ 65% pour la mutation somatique du gène.
Symptômes du rétinoblastome
Certains signes peuvent être perçus en cas de tumeur maligne de la rétine chez le jeune enfant.
– Manifestation d’une leucocorie également connue sous le nom d’œil de chat chez l’enfant. Ce symptôme se caractérise par la présence d’un reflet de couleur blanchâtre au niveau de la pupille de l’œil affecté. Elle peut n’être visible que sous éclairage dans les premières phases de développement de la tumeur et tend à devenir plus nette et permanente à mesure que la pathologie s’aggrave ;
– Strabisme caractérisé par une anomalie du parallélisme des axes visuels. Le deux yeux ne parviennent donc pas à fixer ensemble le même point. À la différence d’un simple strabisme accommodatif, celui-ci n’affecte que l’organe de la vue affecté et est permanent ;
– Aucune douleur n’est en général ressentie dans les premières phases de l’affection pouvant alors cacher la gravité de l’atteinte de la rétine ;
– Inflammation et irritation de l’œil concerné dans des cas plus rares ;
– Décollement de la rétine dans certaines formes de la tumeur ;
– Baisse progressive de l’acuité visuelle aboutissant à la cécité en l’absence de traitements ;
– Sensation de douleur, diminution de l’appétit, vomissement et céphalées dans les cas plus avancés ;
– Apparition d’autres signes à une phase plus tardive tels qu’une buphtalmie (caractérisée par un accroissement du volume oculaire et une proéminence cornéenne), une hétérochromie irienne (apparition d’une autre couleur sur l’iris) ;
– Risque élevé de développer d’autres types de cancer chez les sujets touchés par la forme bilatérale de la tumeur maligne de la rétine.
Traitements classiques du rétinoblastome
La prise en charge de cette tumeur maligne de la rétine doit être débutée le plus tôt possible pour prévenir la survenue de diverses complications et assurer la conservation de l’œil affecté. Parmi tous les traitements utilisés pour soigner cette pathologie, il y a notamment :
– Le laser : L’utilisation du laser est préconisée dans les cas d’atteinte de faible envergure. Elle est pratiquée sous anesthésie générale de l’enfant ;
– La cryoapplication : Le recours à la cryoapplication se fait dans les formes de tumeurs de petites dimensions localisées sur la partie périphérique rétinienne. Trois sessions peuvent alors être nécessaires pour le traitement de chaque affection tumorale (2) ;
– La chimiothérapie : Certaines études avancent la nécessité d’une chimiothérapie notamment en cas de dissémination du cancer ou dans les formes très évoluées avant de passer à un traitement local (3) ;
– L’énucléation : Dans les cas de tumeurs de grande volume, l’énucléation peut être la seule option, une intervention radicale qui consiste à extirper le globe oculaire.
Le traitement de cette maladie relève uniquement de la compétence médicale. Il est donc impératif de consulter un médecin si ces symptômes se présentent.
Références
(1) Rétinoblastome, Encyclopédie de la vue, Syndicat National des Ophtalmologistes de France
(2) Zografos L., Chapitre rétinoblastome, Rapport de la société française d’ophtalmologie, 2002.
(3) Chan HS, et al, Chemotherapy for Retinoblastoma., Ophthalmol Clin North Am. 2005.


